| Editorial |
In
1969, McCully first proposed that homocysteine causes atherosclerosis.1
His hypothesis was based on the finding of atherosclerotic plaque at autopsies
of young people with homocystinuria. This hypothesis was later modified
to include a broader population, positing that mild hyperhomocysteinemia
caused by dietary deficiencies of the vitamin cofactors required for the
metabolism of homocysteine — folic acid, vitamin B12, and vitamin
B6 — is a risk factor for atherothrombosis. In developed countries,
these vitamins are partially removed from foods during processing,2 and
typical diets are rich in the precursor amino acid methionine (which is
derived from animal proteins). These conditions result in elevated homocysteine
concentrations.
Although at first not generally accepted, epidemiologic studies conducted
over the past 25 years have provided ample support for the association
of mild hyperhomocysteinemia with an elevated risk of atherothrombosis.
In a meta-analysis of prospective observational studies of first events,
the members of the Homocysteine Studies Collaboration concluded that a
25 percent reduction in the serum homocysteine concentration (a reduction
of approximately 3 µmol per liter) is associated with an 11 percent
lower risk of ischemic heart disease (odds ratio, 0.89; 95 percent confidence
interval, 0.83 to 0.96) and a 19 percent lower risk of stroke (odds ratio,
0.81; 95 percent confidence interval, 0.69 to 0.95).3 The results of prospective
studies of recurrent cardiovascular events are more consistent than those
for first events; they show in general that the hazard ratio for a recurrent
event increases by 16 percent with each increase of 5 µmol per liter
in the serum homocysteine concentration.4
The independent risk of cardiovascular events conferred by mildly elevated
serum homocysteine levels and the association of elevated levels with
a deficiency of folic acid and vitamin B12 have offered a unique target
for preventive approaches. The metabolism of homocysteine is complex.
In hepatic cells, it involves transsulfuration (by means of the vitamin
B6–dependent rate-limiting enzyme cystathionine -synthase) to cystathionine
and thence to cysteine; in nonhepatic cells, the principal pathway is
remethylation to methionine. Methionine synthesis is based on the folic
acid–dependent and vitamin B12–dependent activity of methionine
synthase or the betaine-dependent activity of betaine–homocysteine
methyltransferase.
Several large, prospective trials have been initiated over the past five
years to study the consequences on cardiovascular events of lowering serum
homocysteine concentrations with the use of folic acid, vitamin B12, and
vitamin B6. The ease of administration of these inexpensive, naturally
occurring cofactors has offered a straightforward approach to testing
the homocysteine hypothesis. Data from animal models and small trials
in humans involving surrogate end points — including measurements
of endothelial function and markers of oxidant stress and inflammation
and their responses to folic acid (and vitamin B12) — have yielded
reasonably consistent results.5,6,7,8 Some study results have differed
depending on the dose and duration of folic acid therapy and its independent
benefit with regard to vascular function.9,10 Nevertheless, this overall
body of work has provided a credible basis for the design of the main
trials.
Three of these prospective trials of the effects of homocysteine-lowering
therapy on recurrent cardiovascular events among subjects with known cardiovascular
disease have now been completed.11,12,13 In the Vitamin Intervention for
Stroke Prevention (VISP) trial,11 two groups of patients with stroke (3680
patients in total) were treated with different daily doses of folic acid,
vitamin B12, and vitamin B6; after two years, there was a dose-dependent
reduction in homocysteine concentration but no significant difference
in the rates of vascular events between the two groups.
The results of the Norwegian Vitamin (NORVIT) trial12 and the Heart Outcomes
Prevention Evaluation (HOPE) 2 trial,13 both reported in this issue of
the Journal, are similar. The NORVIT trial was a study of secondary prevention
involving 3749 patients who had had an acute myocardial infarction and
who were treated daily with folic acid, vitamin B12, and vitamin B6; folic
acid and vitamin B12; vitamin B6 alone; or placebo. After a median follow-up
of 40 months, despite a 27 percent lowering of the mean total homocysteine
concentration from the baseline value among those treated with folic acid
and vitamin B12, there was no significant effect of folic acid and vitamin
B12 on the risk of the composite primary end point of recurrent myocardial
infarction, stroke, or sudden death from coronary artery disease. There
was, however, a near-significant trend toward more myocardial infarctions,
as well as a marginally significant trend toward fewer strokes, among
patients receiving folic acid, vitamin B12, and vitamin B6 than among
those receiving placebo.
HOPE-2 was a prevention trial involving 5522 patients with vascular disease
or diabetes who were treated daily with a combination of folic acid, vitamin
B12, and vitamin B6 or with placebo for an average of five years. Again,
vitamin treatment was associated with a substantial reduction in plasma
homocysteine concentration but not with a significant reduction in the
risk of the composite primary end point of myocardial infarction, stroke,
or death from cardiovascular causes. In addition, this trial showed a
marginally significant reduction in stroke among the patients receiving
vitamins than among those receiving placebo.
The data are quite consistent among these three similar (but not identical)
patient populations, including patients who had and those who did not
have access to foods fortified with folic acid. Although the vitamin doses
used, the consequences of folic acid fortification on the expected event
rates,14 and the implications of the trend toward lower rates of stroke
could all be debated, the consistency among the results leads to the unequivocal
conclusion that there is no clinical benefit of the use of folic acid
and vitamin B12 (with or without the addition of vitamin B6) in patients
with established vascular disease.
The results also raise two other questions that merit consideration. First,
does the failure of homocysteine-lowering therapy to reduce the rates
of cardiovascular events suggest that the homocysteine hypothesis is incorrect?
And if so, is homocysteine a surrogate for another, metabolically related
species that is the true atherogenic culprit? Although suggested by the
results, affirmative answers to these questions are inconsistent with
the abundant evidence in vitro and in vivo that homocysteine is an atherogenic
determinant that promotes oxidant stress, inflammation, thrombosis, endothelial
dysfunction, and cell proliferation.
Second, if homocysteine is an atherogenic determinant, do the results
of these trials suggest that vitamin therapy has other, potentially adverse
effects that offset its homocysteine-lowering benefit? Three mechanisms
might explain the potential adverse effects of this therapy.
One possible mechanism is that, through its role in the synthesis of thymidine,
folic acid promotes cell proliferation (which is the basis for chemotherapies
that disrupt the methylation cycle). Folic acid may do the same in the
atherosclerotic plaque. This mechanism has been offered as an explanation
for the worsening rates of in-stent restenosis in a recent study of patients
who had undergone angioplasty and were treated with folic acid, vitamin
B12, and vitamin B6.15
Another possible mechanism is based on the relation of homocysteine to
the methylation cycle
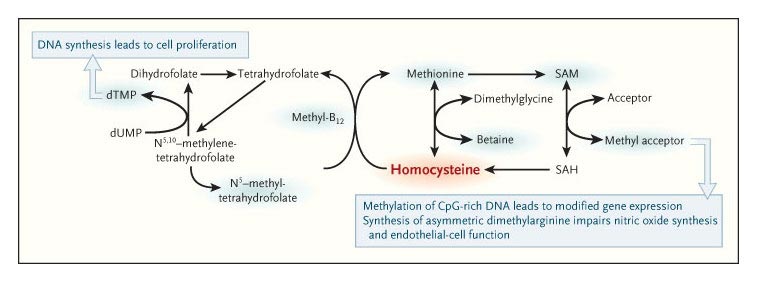
Homocysteine levels are reduced as a result of the enhancement of homocysteine
methylation, which is promoted by folic acid and vitamin B12. This reduction
can be associated with greater overall methylation potential, which can
in turn increase cell proliferation, modify gene expression, and adversely
affect endothelial function. Blue shading indicates methyl-group carriers.
CpG-rich DNA is DNA in which the frequency of the CG sequence is greater
than in other regions. dTMP denotes deoxythymidylate monophosphate, dUMP
deoxyuridylate monophosphate, SAM S-adenosylmethionine, and SAH S-adenosylhomocysteine.
High homocysteine concentrations lead to increased S-adenosylhomocysteine
concentrations: folic acid and vitamin B12 promote the remethylation of
homocysteine to methionine, which in turn lowers S-adenosylhomocysteine
and increases S-adenosylmethionine levels. This latter species is the
sole source of methyl groups for all methylation reactions in the cell.16
As a result of their influence on the steady-state concentrations of these
S-adenosyl derivatives, high homocysteine concentrations are associated
with a reduced methylation potential, whereas folic acid and vitamin B12
increase the methylation potential. The methylation of CpG-rich islands
(short regions of DNA in which the frequency of the CG sequence is greater
than in other regions) in promoter regions of DNA is an epigenetic mechanism
for modulating gene expression. First recognized as a means of silencing
genes during development and of inhibiting carcinogenesis, DNA methylation
also appears to play a role in atherogenesis. Atherogenesis involves local
hypermethylation and hypomethylation of genes, and recently, atherogenic
lipoproteins have been shown to promote DNA hypermethylation in cultured
human macrophages.17,18 Thus, the use of folic acid and vitamin B12 in
the setting of mild hyperhomocysteinemia may alter the methylation potential
in vascular cells, resulting in a change in the cell phenotype that promotes
the development of plaque.
As a third possible mechanism, another important methylation reaction
that can promote atherogenesis, independently of changes in gene expression,
is the methylation of l-arginine to asymmetric dimethylarginine. The latter,
a metabolic product of protein arginine residues, inhibits the activity
of nitric oxide synthase and is associated with an increased risk of vascular
disease. Again, one might predict that by increasing the methylation potential,
treatment with folic acid and vitamin B12 might increase, or at the very
least not change considerably, the concentration of asymmetric dimethylarginine.19
What, then, can we conclude from the results of these trials? Clearly,
folic acid, vitamin B12, and vitamin B6 are not the therapeutic solution
expected, and they do not provide a preventive benefit in patients with
mild hyperhomocysteinemia. The straightforward but incorrect view that
folic acid can decrease homocysteine levels and, thus, reduce the risk
of atherosclerosis effectively may be an unintended consequence of oversimplifying
a complicated metabolic network. Further exploration of the relations
among the intermediates in this metabolic pathway and their association
with atherothrombotic mediators will be needed. Meanwhile, we should consider
alternative approaches to reducing homocysteine concentrations, perhaps
with new methods of enhancing the conversion of homocysteine to cysteine
in the liver or enhancing the urinary excretion of the amino acid.
References
1) McCully KS. Vascular pathology of homocysteinemia: implications
for the pathogenesis of arteriosclerosis. Am J Pathol 1969;56:111-128.
2) Schroeder HA. Losses of vitamins and trace minerals resulting from
processing and preservation of foods. Am J Clin Nutr 1971;24:562-573.
3) Homocysteine Studies Collaboration. Homocysteine and risk of ischemic
heart disease and stroke: a meta-analysis. JAMA 2002;288:2015-2022.
4) Wald DS, Law M, Morris JK. Homocysteine and cardiovascular disease:
evidence on causality from a meta-analysis. BMJ 2002;325:1202-1202.
5) Eberhardt RT, Forgione MA, Cap A, et al. Endothelial dysfunction in
a murine model of mild hyperhomocyst(e)inemia. J Clin Invest 2000;106:483-491.
6) Devlin AM, Arning E, Bottiglieri T, Faraci FM, Rozen R, Lentz SR. Effect
of Mthfr genotype on diet-induced hyperhomocysteinemia and vascular function
in mice. Blood 2004;103:2624-2629.
7) Zhou J, Moller J, Ritskes-Hoitinga M, Larsen ML, Austin RC, Falk E.
Effects of vitamin supplementation and hyperhomocysteinemia on atherosclerosis
in apoE-deficient mice. Atherosclerosis 2003;168:255-262.
8) Woo KS, Chook P, Chan LL, et al. Long-term improvement in homocysteine
levels and arterial endothelial function after 1-year folic acid supplementation.
Am J Med 2002;112:535-539.
9) Doshi SN, McDowell IF, Moat SJ, et al. Folic acid improves endothelial
function in coronary artery disease via mechanisms largely independent
of homocysteine lowering. Circulation 2002;105:22-26.
10) Moat SJ, Doshi SN, Lang D, McDowell IFW, Lewis MJ, Goodfellow J. Treatment
of coronary heart disease with folic acid: is there a future? Am J Physiol
Heart Circ Physiol 2003;287:H1-H7.
11) Toole JF, Malinow MR, Chambless LE, et al. Lowering homocysteine in
patients with ischemic stroke to prevent recurrent stroke, myocardial
infarction, and death: the Vitamin Intervention for Stroke Prevention
(VISP) randomized controlled trial. JAMA 2004;291:565-575.
12) Bønaa KH, Njølstad I, Ueland PM, et al. Homocysteine
lowering and cardiovascular events after acute myocardial infarction.
N Engl J Med 2006;354:1578-1588.
13) The Heart Outcomes Prevention Evaluation (HOPE) 2 Investigators. Homocysteine
lowering with folic acid and B vitamins in vascular disease. N Engl J
Med 2006;354:1567-1577.
14) Bostom AG, Selhub J, Jacques PF, Rosenberg IH. Power shortage: clinical
trials testing the "homocysteine hypothesis" against a background
of folic acid-fortified cereal grain flour. Ann Intern Med 2001;135:133-137.
15) Lange H, Suryapranata H, De Luca G, et al. Folate therapy and in-stent
restenosis after coronary stenting. N Engl J Med 2004;350:2673-2681.
16) Loscalzo J. Adverse effects of supplemental L-arginine in atherosclerosis:
consequences of methylation stress in a complex catabolism? Arterioscler
Thromb Vasc Biol 2003;23:3-5.
17) Zaina S, Lindholm MW, Lund G. Nutrition and aberrant DNA methylation
patterns in atherosclerosis: more than just hyperhomocysteinemia? J Nutr
2005;135:5-8.
18) Lund G, Andersson L, Lauria M, et al. DNA methylation polymorphisms
precede any histological sign of atherosclerosis in mice lacking apolipoprotein
E. J Biol Chem 2004;279:29147-29154.
19) Boger RH, Bode-Boger SM, Sydow K, Heistad DD, Lentz SR. Plasma concentration
of asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide
synthase, is elevated in monkeys with hyperhomocyst(e)inemia or hypercholesterolemia.
Arterioscler Thromb Vasc Biol 2000;20:1557-1564.
|